14. Challenges#
14.1. Introduction#
READ THIS CAREFULLY!
All the challenges in this chapter require you to use many of the learned concepts of this course. They are star-coded to indicate the difficulty level, ranging from one ✲ to five ✲✲✲✲✲. Some challenges have one star in their minimal form, but represent more stars when finished completely. For most of these more difficult challenges you will need to learn to use new libraries, such as GUI toolkits.
You have to create a real program, in a dedicated script, with a main() method that is called from within this construct:
if __name__ == "__main__":
#Start application here
To become a real-world programmer you could and should also start making use of the argparse module to process command-line arguments. When grading your work, these aspects are all taken into account:
Code quality (names, pep-8
There is a minimal amount of top-level code (global variables, code outside functions)
Almost all code resides within small functions. These are well-documented and serve only a single well-defined purpose.
Code efficiency (using right language constructs)
Use of correct build-in functions and libraries
Complexity of the implemented challenge
Use of the
argparsemodule. See here for the official docs, and below for a small demonstration program.
Note that the first challenges are generic; they are not related to (molecular) biology or bio-informatics and thus may be easier to start out with.
Besides the challenges listed below, you are encouraged to think of one yourself. Alternatively, you can look at https://rosalind.info, a nice site offering many challenges related to bioinformatics. Check with the lecturer when you have chosen one if whether it is appropriate as final challenge.
14.1.1. The argparse module#
The argparse module can be used to process command-line (terminal) commands in a standards-compliant manner.
Almost all programs need external input to work correctly, either as data source or program arguments that modify the way the program operates.
For instance, suppose you have a program, my_app.py, that needs to know the username of the user, and some textual input that can come either from the commandline directly, or from a file.
Below is a listing of some examples of how a typical run of this program could look like (I have omitted the python interpreter command tha comes first).
my_app.py --help # shows help on usage
my_app.py -h # same, short form
my_app.py -n Mike -t "This is the input" # gives username and terminal input
my_app.py -n Mike --file data.txt # gives username and file input (long form)
my_app.py -n Mike -f data.txt -v # gives username and file input and runs verbose
When using the argparse module, this is quite easy to implement. It requires two pieces of code: the code defining the possible arguments, and a piece of code processing the actual input (from the sys.argv list).
Here is the definition of possible inputs, all put in a single function:
import argparse
def create_argparser():
parser = argparse.ArgumentParser(
prog = 'my_app.py',
description = 'This program demonstrates the usage of the argparse module.',
epilog = 'If you read this: great - you are a thorough person!')
# required option that takes a value
parser.add_argument('-n', '--name', required = True, help = 'The name of the player')
# optional option that takes a value
parser.add_argument('-t', '--text', required = False, help = 'The text to be processed')
# optional option that takes a value (required = False is the default)
parser.add_argument('-f', '--file', help = 'The file to be processed')
# optional on/off flag that will default to false
parser.add_argument('-v', '--verbose', action='store_true',
help = 'The to have more informative messages during program run')
return parser
Below is the processing of the options defined above. The set options can simply be accessed using the dot operator and the long name of the option: args.option. If they are absent, the test if args.option will fail because its value is None.
def process_commandline(parser):
args = parser.parse_args()
print(f'Welcome, {args.name}')
if args.verbose: # Flag is a bool!
print('Running in verbose mode.')
if args.text: ## Checking whether test has been passed
print(f'Reading text from CL: {args.text}')
elif args.file: ## ..if not, look for a file
print(f'Reading from {args.file}')
else: ## if both are absent, I cannot proceed
print('No input')
return args
Note that -h and --help need not be defined - they are implemented by the argparse module itself!
The main block linking these two looks like this:
if __name__ == '__main__':
parser = create_argparser()
program_args = process_commandline(parser)
Below follow some actual runs, and the output that results from the given command.
~$ myapp.py
usage: my_app.py [-h] -n NAME [-t TEXT] [-f FILE] [-v]
my_app.py: error: the following arguments are required: -n/--name
~$ myapp.py --help
usage: my_app.py [-h] -n NAME [-t TEXT] [-f FILE] [-v]
This program demonstrates the usage of the argparse module.
optional arguments:
-h, --help show this help message and exit
-n NAME, --name NAME The name of the player
-t TEXT, --text TEXT The text to be processed
-f FILE, --file FILE The file to be processed
-v, --verbose The to have more informative messages during program run
If you read this: great - you are a thorough person!
~$ myapp.py -n Mike
Welcome, Mike
No input
~$ myapp.py -n Mike -t "datadatadata" -v
Welcome, Mike
Running in verbose mode.
Reading text from CL: datadatadata
~$ myapp.py -n Mike --file data.txt
Welcome, Mike
Reading from data.txt
Note that this only skims the surface: this example only deals with optional arguments, not positional ones. If you want to be more flexible I suggest you read the docs!
14.2. A number guessing game#
Difficulty level: ✲
Write an application that asks the user to think of a number, and try to guess this number as efficiently as possible.
For instance, program output could be:
Please think of a number between 0 and 10. Press enter when you are ready.
Is the number above 3? [enter y for yes and n for no]
n
Is the number above 1? [enter y for yes and n for no]
y
Is the number 2?
n
The number must be 3!
Think before you start! What would be the most efficient “algorithm” to find the number?
You can upgrade this assignment by making it a GUI application using the Panel library (see the website)
14.3. Anagram maker#
Difficulty level: ✲✲
An anagram is a word or phrase formed by rearranging the letters of a different word or phrase, typically using all the original letters exactly once. For example, the word anagram itself can be rearranged into nag a ram. (source: Wikipedia)
You should ask your user for a word (or word combination or phrase) and also (optionally) how many separate words the anagram may or must comprise. Next, present a list of anagrams. Preferable, these anagrams should be pronouncable by intelligently placing vowels and consonants.
You can upgrade this assignment by making it a GUI application using the Panel library (see the website)
14.4. Hangman word guessing game#
Difficulty level: ✲✲
Hangman is a word-quessing game where one party (in this case the computerprogram you will write) chooses a word that the other party needs to guess. The number of letters in the word is indicated with dashes or underscores.
The guessing party takes turns guessing wich letter will be in the word. If the guessed letter is in the word the dashes are replaced at the correct position(s) with the letter. If the guessed letter is not in the word, a gallow is build piece by piece (one piece for each absent letter). In total, the gallow consists of nine parts, including the hanged.
You can choose to visualize progress using another picture of course. The easiest way is using “ASCII art”, as in this example:
HANGMAN = [
'''
+---+
| |
O |
/|\ |
/ \ |
|
=========''']
There are word lists in the data folder of this repo: dutch_words.txt and english_words.txt.
You can upgrade this assignment by making it a GUI application using the Panel library (see the website)
14.5. Encryption using Caesar cipher#
Difficulty level: ✲✲ to ✲✲✲
Caesar’s cipher is one of the simplest and most widely known encryption techniques. Each letter in a text is replaced by a letter some fixed number of positions down (or iup) the alphabet. For example, with a left shift of 3, D would be replaced by A, E would become B, and so on. The method is named after Julius Caesar, who used it in his private correspondence (see Wikipedia).
It is your task to create such a cipher, where the program takes two command-line arguments. The first one is the shift (left being negative) and the second a filename or a literal text.
You can upgrade this assignment by making it a GUI application using the Panel library (see the website)
14.6. Blackjack (Eenentwintigen)#
Difficulty level: ✲✲✲
Implement the game Blackjack (Eenentwintigen in Dutch - Wikipedia) where the user can play against your app.
You can just make it a terminal app, or if you like the challenge (upgrade of difficulty), you can make a user interface using the Panel library (see the website) or any other GUI toolkit, such as TKinter (see here).
14.7. Wordle / Lingo game#
Difficulty level: ✲✲✲ to ✲✲✲✲
Lingo was one of the longest-running tv game shows. In this game the player needs to guess a word of a fixed length (usually between 5 and 7 letters). It looks for instance like this:

It is you challenge to create such a game.
Configurable (through interface or command-line arguments) should at least be the word length (with default of 5), the language (use a default that suits you) and -optionally- the number of guesses a user can take, with a default of 6.
After a guess, the user receives information about the letters on correct positions, letters at wrong positions, and wrong letters.
There are word lists in the data folder of this repo that you can use to randomly pick words: dutch_words.txt and english_words.txt.
Many ways exist to make this challenge really challenging. For instance, you can create a GUI (there are several libraries for that purpose available in Python but I suggest you use TkInter - see here) or the Panel library (see the website).
14.8. A puzzle game#
Difficulty level: ✲✲✲ to ✲✲✲✲✲
Especially older people will probably know the windows Minesweeper game. It looks like this:

I have programmed a slightly different version of it using Python and a GUI framework called TKinter:
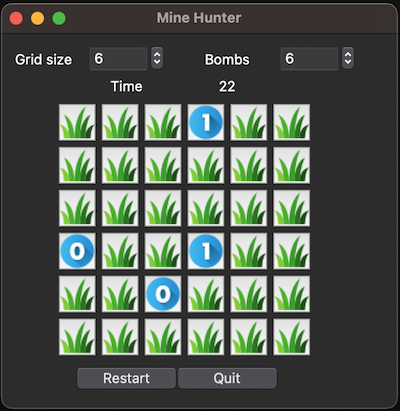
The app code is in the repo of this book here (in folder gui_demo).
You can download that entire repository as zip archive (use the button Code).
Study the code, and read or watch some tutorials.
Next, head over to https://www.puzzle-tents.com/ and choose a game you would like to re-create. Implement that game using python and tkinter.
14.9. Finding palindromes#
Difficulty level: ✲ to ✲✲✲✲
A palindrome is a word, phrase, or sequence that reads the same backwards as forwards, e.g. “madam” or “nurses run”. In biological sequence analysis palindromes are often important protein-binding DNA elements. They are sometimes literal palindromes, as in “GGATTAGG”, or complementary palindromes, where the same sequence is present in the reverse complement strand of the double helix, as in GGAATTCC:
5'-GGAATTCC-3'
3'-CCTTAAGG-5'
Start by writing a single function that determines whether a given input string is a palindrome.
Next, write a function that looks for a palindrome with a minimal length within a given input string, where the rest of the string is not important.
Extend this to processing a word list passed through the command-line. There are two such lists to be found in the
datafolder of this repo:dutch_words.txtandenglish_words.txtApply yourself to molecular biology: create a program that looks for complementary -or optionally non-complementary i.e. literal- palindromes in a given DNA sequence (from file or from the command line). The sequence below contains at least 2 palindromes of differing lengths. Challenges could be: Find all 6-nucleotide palindromes, or What is the longest palindrome in the human genome?
--><-- ----------><----------
5'-AGAGGATCCCTCCCATATATGCGGCGATCGCCGCATATCC-3'
Note that if there is a spacer allowed between the two “legs” of the palindrome, this makes the problem much harder to solve.
You can upgrade this assignment by making it a GUI application using the Panel library (see the website)
14.10. Create a data dashboard#
Difficulty level: ✲✲ to ✲✲✲✲
Create an app that receives a file with structured data (columns) as input. The app then reads this file in a streaming manner and reports statistics of the file contents. The first three of these are mandatory, the subsequent features are optional and increase the level of this challenge.
Dimensions of the dataset (rows/comuns)
Type of data in the columns
Descriptive statistics of each column;
if numeric: a five-number summary plus the mean
if character: the number of unique values and a listing of frequencies of these if the number of unique values is below 10
A pairwise correlation matrix of the numeric columns (if less than 6 or so). For this you will need to use something like numpy, see here
Embed this in a GUI; there are several libraries for that purpose available in Python but I suggest you use the Panel library (see the website). Or, if you prefer some extra supercharged challenge: TkInter, PyQt5 or PySimpleGUI - see here.
Be creative; and impress yourself and your teacher.
14.11. Fasta parser and search tool#
Difficulty level: ✲✲✲✲
Given the Fasta sequence format, create a parser for files of this type.
Next, create an executable that can be used for these use cases (not all need to be implemented to have a passable project):
FastaQuery.py --help– shows informative help/usage information.FastaQuery.py –infile <INFILE> --summary– Creates a textual summary of the parsed file: number of sequences and average lengthFastaQuery.py –infile <INFILE> --word-freqs --length <LENGTH>– Reports word frequencies of the given length in the sequences, (optionally) with as statistical analysis of their expected and actual frequency.FastaQuery.py –infile <INFILE> --fetch_id <ID>– Returns the sequence with this ID, in Fasta format.FastaQuery.py –infile <INFILE> --search <string>– Returns the sequences with this string somewhere in the header, in Fasta format.FastaQuery.py –infile <INFILE> --find_sites <DNA SEQ WITH IUPAC CODES>– Lists the sequences and locations of all the sites where the DNA pattern is found: position and actual sequence and (if relevant) the element in which it resides.
Fasta files of whole genomes (DNA or protein!) can be downloaded from https://www.ncbi.nlm.nih.gov/datasets/genome/.
Simply search something like Bacillus subtilis and click on an entry. You can then choose to download sequences in different formats.
Choose “Sequence and Annotation” (gbff format).
Alternatively, you can look for gene families using for instance protein; e.g. look for FHIT. Or do a BLAST and download all sequences as Fasta.
14.12. Sequence Profiler#
Difficulty level: ✲✲✲ to ✲✲✲✲
This challenge revolves around multiple sequence alignment of DNA sequences, like the ones below:
ungapped
A T T G C C A T T
A T G G C C A T T
A T C T C A A T T
A T C T T C C T T
A C T G A C C T C
gapped
A T T G C C A T T
A T - G C C A T T
A T - T C A A T T
A T C T T C C T T
A C T G A C C T C
Given such an aligned set of sequences, provided in an input file, it is your task to build a profile using the IUPAC ambiguity codes and report this to the user. In the above example, the first profile would look something like this:
A Y B K H M M T Y
Supercharge your challenge: using such a sequence profile, can you implement the functionality to search a sequence (set) for the presence of this, or any other given profile?
You can also upgrade this assignment by making it a GUI application using the Panel library (see the website)
14.13. FastQ analyser#
Difficulty level: ✲✲✲ to ✲✲✲✲
Below is an example of FastQ sequences, exactly as you would find them in a file generated by a high-throughput sequencer such as Illumina HiSeq:
@HWI-962:64:D0BNFACXX:7:1101:1227:2119 1:N:0:GATCAG
GACATGTTAGATCACTCTTTGGGATGTGTTATCTTGGTGTGTGAAATGAATGTGGGAATTGTTGCACAATTTTTTTTGTTTGAATACTTAATTTGTTGAN
+
CCCFFFFFHHHHHJJJJJJJJJJHIJHHIJIJIJJJJGHDHHIIJIJJFIIJHIIJIJJJJJJEHJJIGIJJJJHDDDDDDDDDDEDDDECEDEDEDB@#
@HWI-962:64:D0BNFACXX:7:1101:1176:2147 1:N:0:GATCAG
CTTTGATGTTGACCGAAAAGATTCAGGAAAGAAGTCTGCCTGAATCTCGAACGAGAAGGACAGTATATCACCTCTATCATTCGATGAGTAATTGGAAAAN
+
CCCFFFFFHHHHHIJJJIJEIIJJGIIIIJJIJGIJIGHIIIJJJHHHIEGGIIGGHJHF@HFFFFEFBCCECEEDDCDDEDEDD@ACACC@DA@CDDD#
This format is described here.
It is your task to read the data in a FastQ file provided by the user, read and process the sequences therein, and report on various quality aspects of the sequences. These can be
number of sequences
length (distribution) of sequences
G/C content
number of ‘N’ nucleotides per position, per sequence
average quality
per-base quality
sliding-window quality
K-mer content
Supercharge your challenge: add a “trimming” and “filtering” option to the app.
You can upgrade this assignment by making it a GUI application using the Panel library (see the website)
14.14. Chemistry toolbox#
Difficulty level: ✲✲ to ✲✲✲✲✲
Create an app that implements some of the use cases listed below.
pH Calculation: Calculate the pH of a solution given its concentration of hydrogen ions (H⁺). The user should input the H⁺ concentration, and the program should output the pH value. You can use the formula pH = -log[H⁺].
Molecular Weight: Calculate the molecular weight of a chemical compound. The user should input the chemical formula, and the program should output the total molecular weight. You can use a dictionary to store the atomic weights of each element.
Electron Configuration Generator: Generate the electron configuration of an element based on its atomic number. The program should output the distribution of electrons in each energy level according to the Aufbau principle.
Balancing Chemical Equations: The user inputs an unbalanced chemical equation, and the program outputs the balanced equation. You can represent the equation as a matrix and solve it using linear algebra or implement an algorithm like Gauss-Jordan elimination.
Ideal Gas Law Calculator: Calculate various properties of an ideal gas using the ideal gas law equation (\(PV = nRT\)). The user inputs the values of pressure, volume, temperature, and the number of moles, and the program calculates the missing parameter or performs other related calculations.
Chemical Reaction Simulator: Build a simple chemical reaction simulator that allows the user to input reactants and their quantities, then predicts the products of the reaction based on predefined reaction rules. You can represent chemical reactions as dictionaries or classes and apply basic stoichiometry principles.
Can you think of some interesting functionality yourself? I am a simple biologist…
14.15. Protein analysis tool#
Difficulty level: ✲✲✲
Proteins consist of long chains of amino acids called polypeptides. These constituent amino acids define the structure and properties of the protein. Amino acids have several relevant properties:
size
molecular weight
polarity
charge
hydrophobicity
Amino acid properties can be found on many places; here is one.
Given a protein sequence file (in Fasta format), read in all sequences within the file and generate (choose which are feasable for you to implement):
A comma-separated values (csv format) file with for each protein some summary statistics:
name
length (in amino acids)
molecular weight
average hydrophobicity
total charge
the frequencies of the 20 amino acids
For each sequence a separate file where, using a sliding window technique. The name of the protein is the file name plus
.csvas extension. The file has, also in csv format, four columns of data:sequence position
average hydrophobicity for the current window
average charge for the current window
average polarity for the current window (The second part is harder)
As a command-line arguments this tool should accept the input multifasta file, the sliding window size (with a default of 8) and an output folder to write all results to (default to same directory as input file).
14.16. Genbank query tool#
Difficulty level: ✲✲✲✲
Given the GenBank format (assume only one DNA sequence resides in a single file – if there are more, ignore these and report this to the user).
Create a parser for files of this type, extracting of all Features only these ones:
Source/organism
CDS/coordinates
CDS/product
CDS/db_xref
CDS/translation
Gene/coordinates
Gene/gene
Sequence
Create an appropriate data model. Object-oriented may be a nice challenge here but this is not required.
Create an executable that can be used for these use cases (not all need to be implemented to have a passable project):
GenBankReader.py --helpshows informative help/usage information.GenBankReader.py --infile <INFILE> --summary– Creates a textual summary of the parsed file: length of the sequence, number and types of features. Average lengths of the genes, number on forward strand, number on reverse strand. Maybe you can think of some more interesting features?GenBankReader.py --infile <INFILE> --fetch_gene <GENE NAME (-PATTERN)>– Returns nucleotide sequences of the genes that match the gene name pattern, in Fasta format.GenBankReader.py --infile <INFILE> --fetch_cds <PRODUCT NAME (-PATTERN)>– Returns the amino acid sequences of the CDSs that match the product name pattern, in Fasta format.GenBankReader.py --infile <INFILE> --fetch_features <COORDINATES>Returns all features between the given coordinates.GenBankReader.py --infile <INFILE> --find_sites <DNA SEQ WITH IUPAC CODES>– Lists the locations of all the sites where the DNA pattern is found: position and actual sequence and (if relevant) the element in which it resides.
You may want to implement other features that you think are also or more interesting.
Genbank files can be downloaded from https://www.ncbi.nlm.nih.gov/datasets/genome/. Simply search something like Bacillus subtilis and click on an entry. You can then choose to download sequences in different formats. Choose “Sequence and Annotation” (gbff format).
14.17. GFF query tool#
Difficulty level: ✲✲✲✲
Given the GFF3 sequence format (with or without sequences appended in-file; if not appended, a separate sequence file may be provided), create a parser for files of this type extracting all annotations. Create an appropriate data model. Next. create an executable that can be used for these use cases (not all need to be implemented to have a passable project):
GffQuery.py --help– shows informative help/usage information.GffQuery.py –infile <INFILE> --summary– Creates a textual summary of the parsed file: length of the sequence, number and types of annotations. Average lengths of the genes, number on forward strand, number on reverse strand. Maybe you can think of some more interesting features?GffQuery.py –infile <INFILE> --fetch_id <ID>– Returns the nucleotide sequence of the element with this ID, in Fasta format.GffQuery.py –infile <INFILE> --fetch_features <FEATURE_TYPE_>– Returns the nucleotide sequence of all the elements of this type, in Fasta format.GffQuery.py –infile <INFILE> --fetch_region <COORDINATES>– Returns all features between the given coordinates, in gff format.GffQuery.py –infile <INFILE> --find_sites <DNA SEQ WITH IUPAC CODES>– Lists the locations of all the sites where the DNA pattern is found: position and actual sequence and (if relevant) the element in which it resides.
GFF files can be downloaded from https://www.ncbi.nlm.nih.gov/datasets/genome/. Simply search something like Bacillus subtilis and click on an entry. You can then choose to download sequences in different formats. Choose “Sequence and Annotation” (gbff format).
14.18. k-Means clustering#
Difficulty level: ✲✲✲✲
The k-Means clustering algorithm is used much in data science to group data in an unsupervised manner. See Wikipedia for details.
For this assignment you should implement this algorithm in a program that takes two command-line arguments. The first is a file with the input data and the second should be an optional argument for the k-parameter which defaults to 3.
The output should be a file holding a copy of the input data, but with an extra column appended indicating the cluster number in which the example is put. For instance, given this input, a random sample from the famous “Fisher’s iris” dataset:
Sepal.Length Sepal.Width Petal.Length Petal.Width Species
6.2 3.4 5.4 2.3 virginica
6.7 3.1 4.7 1.5 versicolor
5.8 2.7 5.1 1.9 virginica
4.8 3.0 1.4 0.1 setosa
4.8 3.4 1.9 0.2 setosa
6.5 3.0 5.8 2.2 virginica
5.6 3.0 4.5 1.5 versicolor
4.9 3.1 1.5 0.1 setosa
could be clustered (using only the numerical columns!) as follows:
Sepal.Length Sepal.Width Petal.Length Petal.Width Species cluster
6.2 3.4 5.4 2.3 virginica 1
6.7 3.1 4.7 1.5 versicolor 2
5.8 2.7 5.1 1.9 virginica 1
4.8 3.0 1.4 0.1 setosa 3
4.8 3.4 1.9 0.2 setosa 3
6.5 3.0 5.8 2.2 virginica 2
5.6 3.0 4.5 1.5 versicolor 2
4.9 3.1 1.5 0.1 setosa 3
With only one mismatch between the Species and cluster columns.
14.19. Implement neighbor joining clustering#
Difficulty level: ✲✲✲✲
See Wikipedia for details. You could employ several strategies. The first is classical joining where you end up with a structure with all nodes connected. You can also force the algorithm to stop when a set number of clusters has been formed. This makes it comparable to k-Means but without the random seeding (thus deterministic).
14.20. Create a Game of Life simulation app#
Difficulty level: ✲✲✲✲✲
The Game of Life or Conway’s Game of Life is a simulation of a simple growth experiment. It follows a few simple rules deciding whether a cell (an element in a 2D grid) will live or die. The challenge here is to visualize the simulation so you will need to work with a Graphical User Interface (GUI) toolkit. There are several libraries for that purpose available in Python but I suggest you use TkInter, PyQt5 or PySimpleGUI - see here.